Jump to: Current Research Projects
The Kinetoplastida
Trypanosomes are unicellular protozoan parasites in the group kinetoplastida, which cause animal and Human African Trypanosomiasis (HAT, Sleeping sickness), American trypanosomiasis (Chagas disease), and Leishmaniasis (cutaneous and visceral forms). Collectively, this groups affects more than 1 billion people worldwide and directly contributes to poverty and socioeconomic disparities.
The kinetoplastida, named for their mitochondrial DNA containing sub-organelle (the kinetoplast), diverged from the eukaryotic lineage more than 500 million years ago, resulting in highly divergent genomes from mammals and common genetic models. Approximately, 50% of trypanosome proteins are annotated as hypothetical genes of unknown function. Rather than view this as a limitation, the Hovel-Miner lab views this knowledge gap as an opportunity to uncover novel solutions to functional biology challenges.
To unlock the mysteries of Trypanosomatid genomes, we have developed state-of-the-art forward genetic tools in the African trypanosome (T. brucei) that enable the identification of genes that promote specific phenotypes in these parasites. While any screenable phenotype could be evaluated using our (publicly available) tools, we have a specific focus on identifying proteins and pathways that promote drug resistance.
African trypanosomes
Trypanosomes are a large group of unicellular protozoan parasites infecting a wide range of mammalian hosts, including humans and domestic animals. Most of these parasites are transmitted by insects, and some species can cause severe diseases in both humans and animals. In Africa, trypanosomes inflict a heavy socioeconomic burden due to their detrimental effect on human welfare and agricultural development.

The species Trypanosoma brucei, which is transmitted by tsetse flies, is responsible for epidemics of the fatal human disease sleeping sickness (human African trypanosomiasis) in Sub-Saharan Africa. Sleeping sickness occurs in 36 African countries and causes the second highest mortality rate after HIV/AIDS in some parts of the continent. Most of the affected people depend on agriculture, fishing and hunting and live in rural area with limited access to health services. Wild and domestic animals can harbor the human parasite and form a secondary reservoir of infection for tsetse flies under certain conditions. However, the largest economical impact is probably made by trypanosome species affecting cattle by reducing milk and meat production. Animal African trypanosomiasis is also known by the common name Nagana.
Trypanosome life cycle
Infection occurs when an infected tsetse fly bites a human or animal and injects the parasite into the skin. The initially injected short and stumpy form of the parasite enters the lymphatic system and passes into the bloodstream of the mammalian host where it grows into a long and slender form and multiplies. In the bloodsteam, the parasites travel to other body fluids, including the lymph and spinal fluid, and are even able to pass the blood-brain barrier.
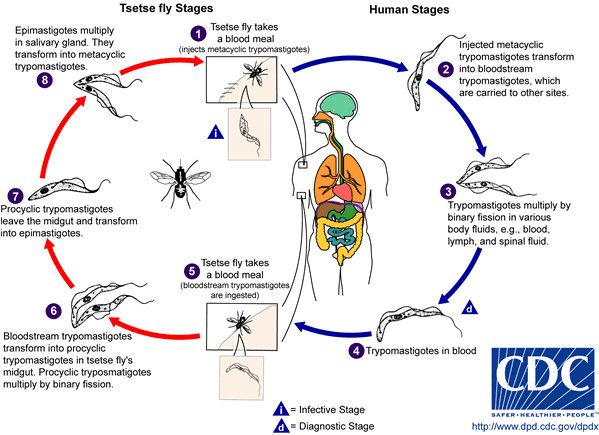
The parasite can only complete its life cycle in the tsetse fly. Tsetse flies become infected when they take up the parasites during a blood meal. The parasites then enter the midgut of the fly where they differentiate and eventually travel to the salivary glands of the fly from where they get injected into the next mammalian host with the saliva on biting.
Current Research Projects
Drug resistance forward genetic screening
American and African trypanosome infections are currently treated with only seven clinically relevant drugs, for which the mechanisms of action are largely unknown. In addition, their limited number makes these drugs susceptible to drug resistance, which is being widely documented. Developing therapies against eukaryotic pathogens is uniquely challenging because, despite their evolutionary divergence, the parasites are inherently more similar to their host than bacteria or viruses, which increases the probability of off-target host toxicity. One strategy for drug design is to identify novel druggable aspects of pathogen biology that differ from the host. However, most anti-trypanosome drugs have been identified empirically based on their parasite-killing without prior knowledge of the drugs' molecular target. We believe that the vast number of uncharacterized hypothetical genes in trypanosome genomes represent an untapped resource of potential drug targets.
To tap this natural resource, our approach is to apply forward genetic screening to existing drugs to both identify their targets as well as additional genes and pathways that can promote drug resistance. In this manner, we will simultaneously determine how the currently utilized drugs function, identify novel targets for optimized drug development and prepare for future resistance outbreaks.
Evaluation of drug induced trypanocidal phenotypes
To better understand drug mechanisms, we are characterizing drug-induced trypanocidal effects using cell biology-based methods. Trypanosomes are distinct from mammalian cells in their cell death processes and specifically do not undergo classical apoptotic pathways. In addition, for trypanocidal drugs, it is usually not known specifically how the drugs induce cell death. Toward our goal of determining drug mechanisms of action, we are establishing measurable trypanocidal phenotypes that result from different drug treatments. For instance, we recently discovered that three drugs in the same category (the nitroaromatic drugs nifurtimox, benznidazole, and fexinidazole) have profoundly different trypanocidal outcomes (doi.org/10.1101/2023.10.09.561529). Elucidating the mechanisms that underlie these outcomes will allow us to determine how antiparasitic drugs kill cells, identify alternative compounds with similar cytotoxic effects, and generate a panel of screenable phenotypes to use in the characterization of genes that promote drug resistance.
Functional characterization of putative resistance genes
Genes that promote survival during drug treatments are identified from our forward genetic screens. This is where the really exciting work begins. We first validate any putative resistance genes to determine if their expression can cause drug resistance or multi-drug resistance. Resistance can arise directly from increased drug target expression, indirectly from increased expression of a pathway that increases the abundance of that target, and indirectly from the expression of proteins that mitigate the damage or stress associated with that drug. The Hovel-Miner Lab is investigating all of these potential outcomes. We believe that genes that potentiate resistance indirectly are as important to understanding drug functions and trypanosome biology as certified drug targets. Through the functional characterization of genes and pathways that promote drug resistance, we will contribute a new understanding of trypanosome functional biology and toward novel drug development strategies.
Trypanosomes as a model for alternative DNA break repair
Human genetic disorders and cancers are frequently associated with defects in DNA break repair. Healthy cells can readily repair DNA damage using either classical non-homologous end joining (c-NHEJ) or homologous recombination (HR). Cancer cells are often defective in these processes and instead rely on alternative forms of DNA break repair, whose pathways and regulatory mechanisms are poorly understood. Trypanosomatids lack essential enzymes of c-NHEJ, which forces them to rely on alt-NHEJ mechanisms more than other eukaryotes. We are using Trypanosoma brucei as a model organism defective in c-NHEJ to identify novel components of alternative DNA break repair pathways in forward genetic screens.